
Computer simulation has become an integral part of the study of the structure and function of biological molecules. For years, parallel computers have been used to conduct these computationally demanding simulations and to analyze their results. These simulations function as a “computational microscope,” allowing the scientist to observe details of molecular processes too small, fast, or delicate to capture with traditional instruments. Over time, commodity GPUs (graphics processing units) have evolved into massively parallel computing devices, and more recently it has become possible to program them in dialects of the popular C/C++ programming languages.
This has created a tremendous opportunity to employ new simulation and analysis techniques that were previously too computationally demanding to use. In other cases, the computational power provided by GPUs can bring analysis techniques that previously required computation on high-performance computing (HPC) clusters down to desktop computers, making them accessible to application scientists lacking experience with clustering, queuing systems, and the like.
This article is based on our experiences developing software for use by and in cooperation with scientists, often graduate students, with backgrounds in physics, chemistry, and biology. Our programs, NAMD18 and VMD10 (Visual Molecular Dynamics), run on computer systems ranging from laptops to supercomputers and are used to model proteins, nucleic acids, and lipids at the atomic level in order to understand how protein structure enables cellular functions such as catalyzing reactions, harvesting sunlight, generating forces, and sculpting membranes (for additional scientific applications, see http://www.ks.uiuc.edu/). In 2007 we began working with the Nvidia CUDA (Compute Unified Device Architecture) system for general-purpose graphics processor programming to bring the power of many-core computing to practical scientific applications.22
Bottom-up Biology
If one were to design a system to safeguard critical data for thousands of years, it would require massive redundancy, self-replication, easily replaceable components, and easily interpreted formats. These are the same challenges faced by our genes, which build around themselves cells, organisms, populations, and entire species for the sole purpose of continuing their own survival. The DNA of every cell contains both data (the amino acid sequences of every protein required for life) and metadata (large stretches of “junk” DNA that interact with hormones to control whether a sequence is exposed to the cell’s protein expression machinery or hidden deep inside the coils of the chromosome).
The protein sequences of life, once expressed as a one-dimensional chain of amino acids by the ribosome, then fold largely unaided into the unique three-dimensional structures required for their functions. The same protein from different species may have similar structures despite greatly differing sequences. Protein sequences have been selected for the capacity to fold, as random chains of amino acids do not self-assemble into a unique structure in a reasonable time. Determining the folded structure of a protein based only on its sequence is one of the great challenges in biology, for while DNA sequences are known for entire organisms, protein structures are available only through the painstaking work of crystallographers.
Simply knowing the folded structure of a protein is not enough to understand its function. Many proteins serve a mechanical role of generating, transferring, or diffusing forces and torques. Others control and catalyze chemical reactions, efficiently harnessing and expending energy obtained from respiration or photosynthesis. While the amino acid chain is woven into a scaffold of helices and sheets, and some components are easily recognized, there are no rigid shafts, hinges, or gears to simplify the analysis.
To observe the dynamic behavior of proteins and larger biomolecular aggregates, we turn to a computational microscope in the form of a molecular dynamics simulation. As all proteins are built from a fixed set of amino acids, a model of the forces acting on every atom can be constructed for any given protein, including bonded, electrostatic, and van der Waals components. Newton’s laws of motion then describe the dynamics of the protein over time. When experimental observation is insufficient in resolving power, with the computer we have a perfect view of this simple and limited model.
Is it necessary to simulate every atom in a protein to understand its function? Answering no would require a complete knowledge of the mechanisms involved, in which case the simulation could produce little new insight. Proteins are not designed cleanly from distinct components but are in a sense hacked together from available materials. Rising above the level of atoms necessarily abandons some detail, so it is best to reserve this for the study of aggregate-level phenomena that are otherwise too large or slow to simulate.
Tracking the motions of atoms requires advancing positions and velocities forward through millions or billions of femtosecond (10−15 second) time steps to simulate nanoseconds or microseconds of simulated time. Simulation sizes range from a single protein in water with fewer than 100,000 atoms to large multicomponent structures of 110 million atoms. Although every atom interacts with every other atom, numerical methods have been developed to calculate long-range interactions for N atoms with order N or N log N rather than N2 complexity.
Before a molecular dynamics simulation can begin, a model of the biomolecular system must be assembled in as close to a typical state as possible. First, a crystallographic structure of any proteins must be obtained (pdb.org provides a public archive), missing details filled in by comparison with other structures, and the proteins embedded in a lipid membrane or bound to DNA molecules as appropriate. The entire complex is then surrounded by water molecules and an appropriate concentration of ions, located to minimize their electrostatic energy. The simulation must then be equilibrated at the proper temperature and pressure until the configuration stabilizes.
Processes at the atomic scale are stochastic, driven by random thermal fluctuations across barriers in the energy landscape. Simulations starting from nearly identical initial conditions will quickly diverge, but over time their average properties will converge if the possible conformations of the system are sufficiently well sampled. To guarantee that an expected transition is observed, it is often necessary to apply steering forces to the calculation. Analysis is performed, both during the calculation and later, on periodically saved snapshots to measure average properties of the system and to determine which transitions occur with what frequency.
If one were to design a system to safeguard critical data for thousands of years, it would require massive redundancy, self-replication, easily replaceable components, and easily interpreted formats. These are the same challenges faced by our genes.
As the late American mathematician R. W. Hamming said, “The purpose of computing is insight, not numbers.” Simulations would spark little insight if scientists could not see the biomolecular model in three dimensions on the computer screen, rotate it in space, cut away obstructions, simplify representations, incorporate other data, and observe its behavior to generate hypotheses. Once a mechanism of operation for a protein is proposed, it can be tested by both simulation and experiment, and the details refined. Excellent visual representation is then needed to an equal extent to publicize and explain the discovery to the biomedical community.
Biomedical Users
We have more than a decade of experience guiding the development of the NAMD (http://www.ks.uiuc.edu/Research/namd/) and VMD (http://www.ks.uiuc.edu/Research/vmd/) programs for the simulation, analysis, and visualization of large biomolecular systems. The community of scientists that we serve numbers in the tens of thousands and circles the globe, ranging from students with only a laptop to leaders of their fields with access to the most powerful supercomputers and graphics workstations. Some are highly experienced in the art of simulation, while many are primarily experimental researchers turning to simulation to help explain their results and guide future work.
The education of the computational scientist is quite different from that of the scientifically oriented computer scientist. Most start out in physics or another mathematically oriented field and learn scientific computing informally from their fellow lab mates and advisors, originally in Fortran 77 and today in Matlab. Although skilled at solving complex problems, they are seldom taught any software design process or the reasons to prefer one solution to another. Some go for years in this environment before being introduced to revision-control systems, much less automated unit tests.
As software users, scientists are similar to programmers in that they are comfortable adapting examples to suit their needs and working from documentation. The need to record and repeat computations makes graphical interfaces usable primarily for interactive exploration, while batch-oriented input and output files become the primary artifacts of the research process.
One of the great innovations in scientific software has been the incorporation of scripting capabilities, at first rudimentary but eventually in the form of general-purpose languages such as Tcl and Python. The inclusion of scripting in NAMD and VMD has blurred the line between user and developer, exposing a safe and supportive programming language that allows the typical scientist to automate complex tasks and even develop new methods. Since no recompilation is required, the user need not worry about breaking the tested, performance-critical routines implemented in C++. Much new functionality in VMD has been developed by users in the form of script-based plug-ins, and C-based plug-in interfaces have simplified the development of complex molecular structure analysis tools and readers for dozens of molecular file formats.
Scientists are quite capable of developing new scientific and computational approaches to their problems, but it is unreasonable to expect the biomedical community to extend their interest and attention so far as to master the ever-changing landscape of high-performance computing. We seek to provide users of NAMD and VMD with the experience of practical supercomputing, in which the skills learned with toy systems running on a laptop remain of use on both the departmental cluster and national supercomputer, and the complexities of the underlying parallel decomposition are hidden. Rather than a fearful and complex instrument, the supercomputer now becomes just another tool to be called upon as the user’s work requires.
Given the expense and limited availability of high-performance computing hardware, we have long sought better options for bringing larger and faster simulations to the scientific masses. The last great advance in this regard was the evolution of commodity-based Linux clusters from cheap PCs on shelves into the dominant platform today. The next advance, practical acceleration, will require a commodity technology with strong commercial support, a sustainable performance advantage over several generations, and a programming model that is accessible to the skilled scientific programmer. We believe that this next advance is to be found in 3D graphics accelerators inspired by public demand for visual realism in computer games.
GPU Computing
Biomolecular modelers have always had a need for sophisticated graphics to elucidate the complexities of the large molecular structures commonly studied in structural biology. In 1995, 3D visualization of such molecular structures required desk-side workstations costing tens of thousands of dollars. Gradually, the commodity graphics hardware available for personal computers began to incorporate fixed-function hardware for accelerating 3D rendering. This led to widespread development of 3D games and funded a fast-paced cycle of continuous hardware evolution that has ultimately resulted in the GPUs that have become ubiquitous in modern computers.
GPU hardware design. Modern GPUs have evolved to a high state of sophistication necessitated by the complex interactive rendering algorithms used by contemporary games and various engineering and scientific visualization software. GPUs are now fully programmable massively parallel computing devices that support standard integer and floating-point arithmetic operations.11 State-of-the-art GPU devices contain more than 240 processing units and are capable of performing up to 1TFLOPs. High-end devices contain multiple gigabytes of high-bandwidth on-board memory complemented by several small on-chip memory systems that can be used as program-managed caches, further amplifying effective memory bandwidth.
GPUs are designed as throughput-oriented devices. Rather than optimizing the performance of a single thread or a small number of threads of execution, GPUs are designed to provide high aggregate performance for tens of thousands of independent computations. This key design choice allows GPUs to spend the vast majority of chip die area (and thus transistors) on arithmetic units rather than on caches. Similarly, GPUs sacrifice the use of independent instruction decoders in favor of SIMD (single-instruction multiple-data) hardware designs wherein groups of processing units share an instruction decoder. This design choice maximizes the number of arithmetic units per mm2 of chip die area, at the cost of reduced performance whenever branch divergence occurs among threads on the same SIMD unit.
The lack of large caches on GPUs means that a different technique must be used to hide the hundreds of clock cycles of latency to off-chip GPU or host memory. This is accomplished by multiplexing many threads of execution onto each physical processing unit, managed by a hardware scheduler that can exchange groups of active and inactive threads as queued memory operations are serviced. In this way, the memory operations of one thread are overlapped with the arithmetic operations of others. Recent GPUs can simultaneously schedule as many as 30,720 threads on an entire GPU. Although it is not necessary to saturate a GPU with the maximum number of independent threads, this provides the best opportunity for latency hiding. The requirement that the GPU be supplied with large quantities of fine-grained data-parallel work is the key factor that determines whether or not an application or algorithm is well suited for GPU acceleration.
As a direct result of the large number of processing units, high-bandwidth main memory, and fast on-chip memory systems, GPUs have the potential to significantly outperform traditional CPU architectures significantly on highly data-parallel problems that are well matched to the architectural features of the GPU.
GPU programming. Until recently, the main barrier to using GPUs for scientific computing had been the availability of general-purpose programming tools. Early research efforts such as Brook2 and Sh13 demonstrated the feasibility of using GPUs for nongraphical calculations. In mid-2007 Nvidia released CUDA,16 a new GPU programming toolkit that addressed many of the shortcomings of previous efforts and took full advantage of a new generation of computecapable GPUs. In late 2008 the Khronos Group announced the standardization of OpenCL,14 a vendor-neutral GPU and accelerator programming interface. AMD, Nvidia, and many other vendors have announced plans to provide OpenCL implementations for their GPUs and CPUs. Some vendors also provide low-level proprietary interfaces that allow third-party compiler vendors such as RapidMind12 and the Portland Group to target GPUs more easily. With the major GPU vendors providing officially supported toolkits for GPU computing, the most significant barrier to widespread use has been eliminated.
Although we focus on CUDA, many of the concepts we describe have analogs in OpenCL. A full overview of the CUDA programming model is beyond the scope of this article, but John Nickolls et al. provide an excellent introduction in their article, “Scalable parallel programming with CUDA,” in the March/April 2008 issue of ACM Queue.15 CUDA code is written in C/C++ with extensions to identify functions that are to be compiled for the host, the GPU device, or both. Functions intended for execution on the device, known as kernels, are written in a dialect of C/C++ matched to the capabilities of the GPU hardware. The key programming interfaces CUDA provides for interacting with a device include routines that do the following:
- Enumerate available devices and their properties
- Attach to and detach from a device
- Allocate and deallocate device memory
- Copy data between host and device memory
- Launch kernels on the device
- Check for errors
When launched on the device, the kernel function is instantiated thousands of times in separate threads according to a kernel configuration that determines the dimensions and number of threads per block and blocks per grid. The kernel configuration maps the parallel calculations to the device hardware and can be selected at runtime for the specific combination of input data and CUDA device capabilities. During execution, a kernel uses its thread and block indices to select desired calculations and input and output data. Kernels can contain all of the usual control structures such as loops and if/else branches, and they can read and write data to shared device memory or global memory as needed. Thread synchronization primitives provide the means to coordinate memory accesses among threads in the same block, allowing them to operate cooperatively on shared data.
The key challenges involved in developing high-performance CUDA kernels revolve around efficient use of several memory systems and exploiting all available data parallelism. Although the GPU provides tremendous computational resources, this capability comes at the cost of limitations in the number of per-thread registers, the size of per-block shared memory, and the size of constant memory. With hundreds of processing units, it is impractical for GPUs to provide a thread-local stack. Local variables that would normally be placed on the stack are instead allocated from the thread’s registers, so recursive kernel functions are not supported.
Analyzing applications for GPU acceleration potential. The first step in analyzing an application to determine its suitability for any acceleration technology is to profile the CPU time consumed by its constituent routines on representative test cases. With profiling results in hand, one can determine to what extent Amdahl’s Law limits the benefit obtainable by accelerating only a handful of functions in an application. Applications that focus their runtime into a few key algorithms or functions are usually the best candidates for acceleration.
As an example, if profiling shows that an application spends 10% of its runtime in its most time-consuming function, and the remaining runtime is scattered among several tens of unrelated functions of no more than 2% each, such an application would be a difficult target for an acceleration effort, since the best performance increase achievable with moderate effort would be a mere 10%. A much more attractive case would be an application that spends 90% of its execution time running a single algorithm implemented in one or two functions.
Once profiling analysis has identified the subroutines that are worth accelerating, one must evaluate whether they can be reimplemented with data-parallel algorithms. The scale of parallelism required for peak execution efficiency on the GPU is usually on the order of 100,000 independent computations. The GPU provides extremely fine-grain parallelism with hardware support for multiplexing and scheduling massive numbers of threads onto the pool of processing units. This makes it possible for CUDA to extract parallelism at a level of granularity that is orders of magnitude finer than is usually practical with other parallel-programming paradigms.
One of the most compelling and successful applications for GPU acceleration has been molecular dynamics simulation, which is dominated by N-body atomic force calculation.
GPU-accelerated clusters for HPC. Given the potential for significant acceleration provided by GPUs, there has been a growing interest in incorporating GPUs into large HPC clusters.3,6,8,19,21,24 As a result of this interest, Nvidia now makes high-density rack-mounted GPU accelerators specifically designed for use in such clusters. By housing the GPUs in an external case with its own independent power supply, they can be attached to blade or 1U rackmount servers that lack the required power and cooling capacity for GPUs to be installed internally. In addition to increasing performance, GPU accelerated clusters also have the potential to provide better power efficiency than traditional CPU clusters.
In a recent test on the AC GPU cluster (http://iacat.uiuc.edu/resources/cluster/) at the National Center for Supercomputing Applications (NCSA, http://www.ncsa.uiuc.edu/), a NAMD simulation of STMV (satellite tobacco mosaic virus) measured the increase in performance provided by GPUs, as well as the increase in performance per watt. In a small-scale test on a single node with four CPU cores and four GPUs (HP xw9400 workstation with a Tesla S1070 attached), the four Tesla GPUs provided a factor of 7.1 speedup over four CPU cores by themselves. The GPUs provided a factor of 2.71 increase in the performance per watt relative to computing only on CPU cores. The increases in performance, space efficiency, power, and cooling have led to the construction of large GPU clusters at supercomputer centers such as NCSA and the Tokyo Institute of Technology. The NCSA Lincoln cluster (http://www.ncsa.illinois.edu/UserInfo/Resources/Hardware/Intel64TeslaCluster/TechSummary/) containing 384 GPUs and 1,536 CPU cores is shown in Figure 1.
GPU Applications
Despite the relatively recent introduction of general-purpose GPU programming toolkits, a variety of biomolecular modeling applications have begun to take advantage of GPUs.
Molecular Dynamics. One of the most compelling and successful applications for GPU acceleration has been molecular dynamics simulation, which is dominated by N-body atomic force calculation. One of the early successes with the use of GPUs for molecular dynamics was the Folding@Home project5,7 where continuing efforts on development of highly optimized GPU algorithms have demonstrated speedups of more than a factor of 100 for a particular class of simulations (for example, protein folding) of very small molecules (5,000 atoms and less). Folding@Home is a distributed computing application deployed on thousands of computers worldwide. GPU acceleration has helped make it the most powerful distributing computing cluster in the world, with GPU-based clients providing the dominant computational power (http://fah-web.stanford.edu/cgi-bin/main.py?qtype=osstats).
HOOMD (Highly Optimized Object-oriented Molecular Dynamics), a recently developed package specializing in molecular dynamics simulations of polymer systems, is unique in that it was designed from the ground up for execution on GPUs.1 Though in its infancy, HOOMD is being used for a variety of coarse-grain particle simulations and achieves speedups of up to a factor of 30 through the use of GPU-specific algorithms and approaches.
NAMD18 is another early success in the use of GPUs for molecular dynamics.17,19,22 It is a highly scalable parallel program that targets all-atom simulations of large biomolecular systems containing hundreds of thousands to many millions of atoms. Because of the large number of processor-hours consumed by NAMD users on supercomputers around the world, we investigated a variety of acceleration options and have used CUDA to accelerate the calculation of nonbonded forces using GPUs. CUDA acceleration mixes well with task-based parallelism, allowing NAMD to run on clusters with multiple GPUs per node. Using the CUDA streaming API for asynchronous memory transfers and kernel invocations to overlap GPU computation with communication and other work done by the CPU yields speedups of up to a factor of nine times faster than CPU-only runs.19
At every iteration NAMD must calculate the short-range interaction forces between all pairs of atoms within a cutoff distance. By partitioning space into patches slightly larger than the cutoff distance, we can ensure that all of an atom’s interactions are with atoms in the same or neighboring cubes. Each block in our GPU implementation is responsible for the forces on the atoms in a single patch due to the atoms in either the same or a neighboring patch. The kernel copies the atoms from the first patch in the assigned pair to shared memory and keeps the atoms from the second patch in registers. All threads iterate in unison over the atoms in shared memory, accumulating forces for the atoms in registers only. The accumulated forces for each atom are then written to global memory. Since the forces between a pair of atoms are equal and opposite, the number of force calculations could be cut in half, but the extra coordination required to sum forces on the atoms in shared memory outweighs any savings.
We expect GPUs to maintain their current factor-of-10 advantage in peak performance relative to CPUs, while their obtained performance advantage for well-suited problems continues to grow.
NAMD uses constant memory to store a compressed lookup table of bonded atom pairs for which the standard short-range interaction is not valid. This is efficient because the table fits entirely in the constant cache and is referenced for only a small fraction of pairs. The texture unit, a specialized feature of GPU hardware designed for rapidly mapping images onto surfaces, is used to interpolate the short-range interaction from an array of values that fits entirely in the texture cache. The dedicated hardware of the texture unit can return a separate interpolated value for every thread that requires it faster than the potential function could be evaluated analytically.
Building, visualizing, and analyzing molecular models. Another area where GPUs show great promise is in accelerating many of the most computationally intensive tasks involved in preparing models for simulation, visualizing them, and analyzing simulation results (http://www.ks.uiuc.edu/Research/gpu/).
One of the critical tasks in the simulation of viruses and other structures containing nucleic acids is the placement of ions to reproduce natural biological conditions. The correct placement of ions (see Figure 2) requires knowledge of the electrostatic field in the volume of space occupied by the simulated system. Ions are placed by evaluating the electrostatic potential on a regularly spaced lattice and inserting ions at the minima in the electrostatic field, updating the field with the potential contribution of the newly added ion, and repeating the insertion process as necessary. Of these steps, the initial electrostatic field calculation dominates runtime and is therefore the part best suited for GPU acceleration.
A simple quadratic-time direct Coulomb summation algorithm computes the electrostatic field at each lattice point by summing the potential contributions for all atoms. When implemented optimally, taking advantage of fast reciprocal square-root instructions and making extensive use of near-register-speed on-chip memories, a GPU direct summation algorithm can outperform a CPU core by a factor of 44 or more.17,22 By employing a so-called “short-range cutoff” distance beyond which contributions are ignored, the algorithm can achieve linear time complexity while still outperforming a CPU core by a factor of 26 or more.20 To take into account the long-range electrostatic contributions from distant atoms, the short-range cutoff algorithm must be combined with a long-range contribution. A GPU implementation of the linear-time multilevel summation method, combining both the short-range and long-range contributions, has achieved speedups in excess of a factor of 20 compared with a CPU core.9
GPU acceleration techniques have proven successful for an increasingly diverse range of other biomolecular applications, including quantum chemistry simulation (http://mtzweb.stanford.edu/research/gpu/) and visualization,23,25 calculation of solvent-accessible surface area,4 and others (http://www.hicomb.org/proceedings.html). It seems likely that GPUs and other many-core processors will find even greater applicability in the future.
Looking Forward
Both CPU and GPU manufacturers now exploit fabrication technology improvements by adding cores to their chips as feature sizes shrink. This trend is anticipated in GPU programming systems, for which many-core computing is the norm, whereas CPU programming is still largely based on a model of serial execution with limited support for tightly coupled on-die parallelism. We therefore expect GPUs to maintain their current factor-of-10 advantage in peak performance relative to CPUs, while their obtained performance advantage for well-suited problems continues to grow. We further note that GPUs have maintained this performance lead despite historically lagging CPUs by a generation in fabrication technology, a handicap that may fade with growing demand.
The great benefits of GPU acceleration and other computer performance increases for biomedical science will come in three areas. The first is doing the same calculations as today, but faster and more conveniently, providing results over lunch rather than overnight to allow hypotheses to be tested while they are fresh in the mind. The second is in enabling new types of calculations that are prohibitively slow or expensive today, such as evaluating properties throughout an entire simulation rather than for a few static structures. The third and greatest is in greatly expanding the user community for high-end biomedical computation to include all experimental researchers around the world, for there is much work to be done and we are just now beginning to uncover the wonders of life at the atomic scale.
![]() Related articles
Related articles
on queue.acm.org
GPUs: A Closer Look
Kayvon Fatahalian and Mike Houston
http://queue.acm.org/detail.cfm?id=1365498
Scalable Parallel Programming with CUDA
John Nickolls, Ian Buck, Michael Garland, and Kevin Skadron
http://queue.acm.org/detail.cfm?id=1365500
Future Graphics Architectures
William Mark
http://queue.acm.org/detail.cfm?id=1365501
Figures
 Figure 1. The recently constructed Lincoln GPU cluster at the National Center for Supercomputing Applications contains 1,536 CPU cores, 384 GPUs, 3TB of memory, and achieves an aggregate peak floating-point performance of 47.5TFLOPS.
Figure 1. The recently constructed Lincoln GPU cluster at the National Center for Supercomputing Applications contains 1,536 CPU cores, 384 GPUs, 3TB of memory, and achieves an aggregate peak floating-point performance of 47.5TFLOPS.
 Figure 2. GPU-accelerated electrostatics algorithms enable researchers to place ions appropriately during early stages of model construction. Placement of ions in large structures such as the ribosome shown here previously required the use of HPC clusters for calculation but can now be performed on a GPU-accelerated desktop computer in just a few minutes.
Figure 2. GPU-accelerated electrostatics algorithms enable researchers to place ions appropriately during early stages of model construction. Placement of ions in large structures such as the ribosome shown here previously required the use of HPC clusters for calculation but can now be performed on a GPU-accelerated desktop computer in just a few minutes.
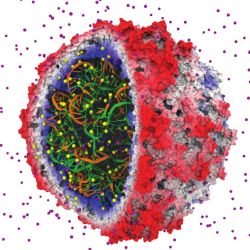
 Figure. An early success in applying GPUs to biomolecular modeling involved rapid calculation of electrostatic fields used to place ions in simulated structures. The satellite tobacco mosaic virus model contains hundreds of ions (individual atoms shown in yellow and purple) that must be correctly placed so that subsequent simulations yield correct results.
Figure. An early success in applying GPUs to biomolecular modeling involved rapid calculation of electrostatic fields used to place ions in simulated structures. The satellite tobacco mosaic virus model contains hundreds of ions (individual atoms shown in yellow and purple) that must be correctly placed so that subsequent simulations yield correct results.



{kind=link}
{kind=link}
{kind=link}
Join the Discussion (0)
Become a Member or Sign In to Post a Comment